Was ist PKU?
PKU bedeutet PhenylKetonUrie und bezeichnet eine angeborene, genetisch bedingte Eiweißstoffwechselstörung.
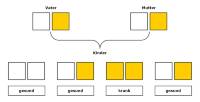
In Deutschland wird ca. jedes 10.000ste Kind mit PKU geboren. Beide Elternteile dieser Kinder sind Träger der Erbanlage und geben diese an ihre Kinder weiter. Da PKU verdeckt - rezessiv - vererbt wird, tritt die Krankheit (mit 25%iger Wahrscheinlichkeit) nur bei den Kindern auf, die von beiden Elternteilen die Anlage zu PKU geerbt haben. Wird die Anlage nur von einem Elternteil weitergegeben, ist das Kind - wie seine Eltern - Träger der Anlage und damit gesund.
Es gibt diese Anlage aber an die folgende Generation mit 50%iger Wahrscheinlichkeit weiter.
Ungefähr jeder 50ste in Deutschland ist Träger der PKU Anlage.
Die Grafik zeigt, wie PKU vererbt wird:
 Phenylalanin (PHE) ist eine von 25 Aminosäuren, den Bausteinen aller Eiweiße. PKU ist ein Enzymdefekt, der den vollständigen Abbau von PHE zu Tyrosin, einer anderen Aminosäure, verhindert. Der Mensch benötigt nur eine kleine Menge PHE täglich, der Rest (ca. 90%) wird normalerweise weiterverstoffwechselt. Nehmen PKU-Betroffene über einen längeren Zeitraum zuviel PHE mit der Nahrung auf, kann es, insbesondere im Wachstumsalter, zu schweren unumkehrbaren körperlichen und geistigen Schäden kommen. Deshalb wird in Deutschland jedes Neugeborene am 5. Lebenstag auf PKU getestet (Fersenblutentnahme durch die Hebamme zuhause oder in der Klinik). Fällt der Test positiv aus, werden die Eltern an das nächst gelegene Stoffwechselzentrum weitervermittelt. Behandlung der PKUJeder Mensch benötigt eine bestimmte Menge PHE am Tag. Diese Menge ist abhängig vom Alter, der Wachstumsgeschwindigkeit, der aktuellen Stoffwechsellage oder dem Trainingszustand (viel sportliche Aktivität / muskelaufbauendes Training hat einen erhöhten Phebedarf zur Folge). Der PKU Patient muss diese Menge kontrolliert zu sich nehmen, da der Körper überflüssiges PHE nicht ausscheiden kann. Das bedeutet, dass der PHE-Gehalt aller Lebensmittel bekannt sein muss, um die aufgenommene PHE-Menge berechnen zu können. Die Analysewerte sind in speziellen Tabellen zusammengefasst und geben den PHE-Gehalt in mg/100g Lebensmittel an. Durch regelmäßige Blutanalysen wird der exakte PHE-Bedarf (PHE-Toleranz) ermittelt. Die Eltern können dann durch Abwiegen der Nahrung die PHE-Aufnahme ihres Kindes steuern. Unsere normalen Lebensmittel enthalten viel zuviel PHE für ein PKU Kind, sodass die Diät auf speziellen eiweißarmen Nahrungsmitteln beruht. Damit durch die eiweißarme Ernährung keine Mangelerscheinungen auftreten, muss eine PHE-freie Aminosäuremischung (z. B. in Pulverform) gegeben werden, die auch alle Vitamine, Mineralien und Spurenelemente enthält. Die Menge dieser Aminosäuremischung berechnet sich aus dem Alter und dem Körpergewicht des Kindes. Aus all diesen Gründen ist es wichtig, die Entwicklung eines PKU-Kindes durch ein Stoffwechselzentrum begleiten und überwachen zu lassen. So ist sicher gestellt, dass die Ernähung und das Zuführen der richtigen Menge an Aminosäuremischung den sich laufend ändernden Bedürfnissen des Kindes angepasst wird. Wird die Diät strikt eingehalten, entwickeln sich PKU-Kinder völlig normal. Das stellt für alle Beteiligten eine große Herausforderung dar, es ist eine zusätzliche Erziehungsleistung zu erbringen. Den Kindern selbst wird viel Disziplin und Einsicht abverlangt. Ärzte und Diätassistenten müssen eine kontinuierliche Betreuung und Begleitung sicherstellen. |